




Una de las cosas más importantes que deben tener en cuenta los desarrolladores de software médico y apps sanitarias son las regulaciones europeas de software médico que les son de aplicación, así como las leyes de protección de datos. En el presente artículo nos centramos en los requerimientos legales que tiene el software de salud en europa, y en este otro artículo encontrarás información respecto al Reglamento General de Protección de Datos (RGPD), la Ley Orgánica de Protección de datos y Garantía de Derechos Digitales (LOPD-GDD) y la Ley de Autonomía del Paciente.

Podemos definir el software médico como el proceso de diseño y desarrollo de tecnologías médicas, que suele darse cuando los investigadores encuentran la posibilidad de desarrollar una tecnología para una necesidad no cubierta. Hallado el nicho, la idea se conceptualiza en uno o diferentes productos.
En una fase temprana del desarrollo de software sanitario suelen buscarse la soluciones más funcionales. ¿La solución es mejor que esta otra tecnología? ¿Quizás es mejor que esta otra solución terapéutica? ¿Tal vez es más rápida?
Una vez está definido el concepto de producto o productos, el objetivo final será producir el software para que sea lanzado al mercado. Si bien no es el objetivo de esta entrada, en este otro artículo encontrarás una guía paso a paso para desarrollar software sanitario que pueda ser exitoso en la industria de la salud.

Desde la conceptualización del producto ya resulta necesario tener en cuenta las regulaciones del software médico. Hay que asegurarse de que el producto que se pretende desarrollar, tal y como se está definiendo, quede bien enmarcado según las normativas y las regulaciones del software sanitario. ¿Qué requisitos legales le aplican, y a qué marco legal hay que someterse? ¿Cuál es el alcance regulatorio del producto que tenemos entre manos?
Lo primero que debemos preguntarnos es si el software que planeamos desarrollar realmente se enmarca como un producto sanitario, ya ue no todos los softwares y apps de salud quedan enmarcados bajo la categoría de software médico.
Simplificando, para que el software pueda ser considerado como médico debe haber un beneficio para un individuo concreto, es decir, que el software médico tome una decisión terapéutica para una persona a nivel individual. Además, para que sea considerado software médico, tiene que estar realizando funciones complejas sobre los datos que se han recabado.
El Marcado CE significa “Conformidad Europea” y es una marca europea que se utiliza para determinados productos industriales, entre otros los productos médicos. Está amparada bajo la Directiva 93/68/CEE.
El proceso regulatorio del software médico debe realizarse a través de los siguientes pasos:
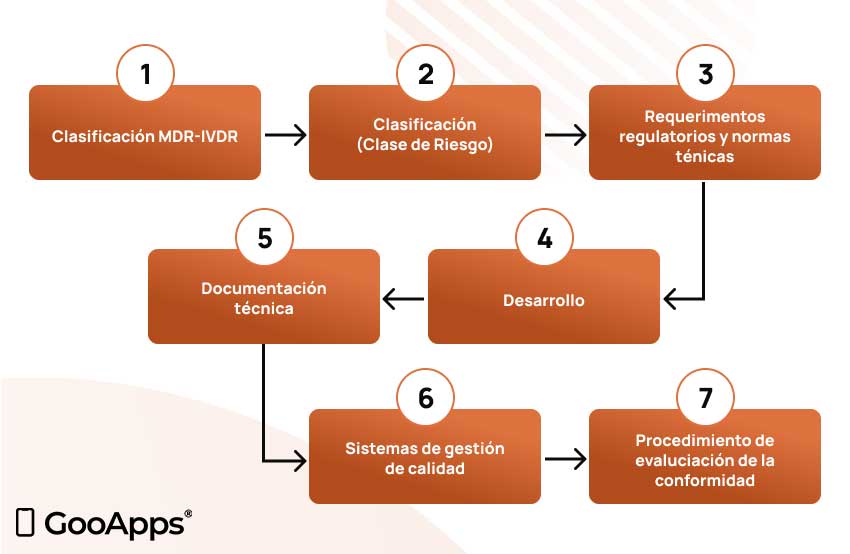
El principal objetivo de las normativas y regulaciones europeas es garantizar que los productos sean seguros para usuarios y/o pacientes, y en segundo plano, que sean funcionales. Por este motivo, se categorizan en función del riesgo que entrañan los posibles errores que puedan derivarse de su uso.
Hay una serie de reglas de clasificación que deben ser revisadas de forma sistemática, según el uso previsto que tiene el producto que estamos desarrollando. En este sentido, existen dos regulaciones europeas principales de aplicación en los productos médicos. Hablamos de la Medical Devices Regulation (EU) 2017/745 (MDR), además de la In Vitro Medical Devices Regulation (EU) 2017/746 (IVDR).
Un producto sanitario para diagnóstico in vitro según la 2017/746 (IVDR) es cualquier producto sanitario que consista en un reactivo, producto reactivo, calibrador, material de control, estuche de instrumental y materiales, instrumento, aparato, equipo o sistema, utilizado solo o en combinación con otros, destinado por el fabricante a ser utilizado in vitro para el estudio de muestras procedentes del cuerpo humano, incluidas las donaciones de sangre y tejidos, sólo o principalmente con el fin de proporcionar información relativa a:
Por lo tanto, un producto sanitario que esté en contacto con el cuerpo humano, como puede ser el caso de un wearable, no entra en la categoría de IVDR. Para que sea un IVDR tiene que estar trabajando con muestras de forma ex vivo.
Una vez determinada la categoría del software médico desarrollado (MDR o IVDR), determinaremos su clasificación en cuanto a la clase de riesgo que le resulta de aplicación. Esta clase de riesgo viene determinada por las consecuencias que pueden tener los posibles errores del producto desarrollado.
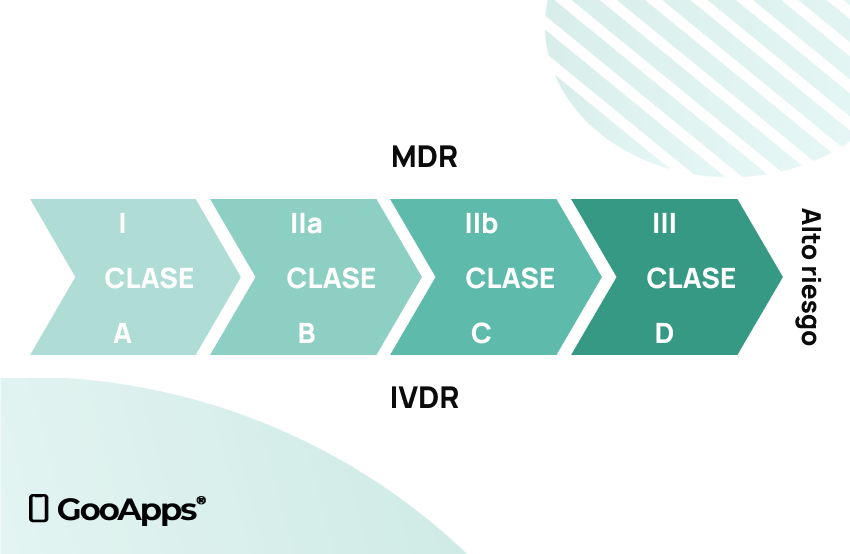
Los programas informáticos destinados a proporcionar información que se utiliza para tomar decisiones con fines terapéuticos o de diagnóstico se clasifican en la clase IIa, salvo si estas decisiones tienen un impacto que pueda causar:
Los programas informáticos destinados a observar procesos fisiológicos se clasifican en la clase IIa, salvo si se destinan a observar parámetros fisiológicos vitales, cuando la índole de las variaciones de dichos parámetros sea tal que pudiera dar lugar a un peligro inmediato para el paciente, en cuyo caso se clasifican en la clase IIb.
Todos los demás programas informáticos se clasifican en la clase I.
Teniendo en cuenta el uso previsto de un producto, y en el caso de que puedan aplicar varias reglas o si, para una misma regla, son aplicables diferentes subreglas, siempre aplicarán la regla y subregla más estrictas que den lugar a la clasificación más elevada.
En el caso de un software que monitorice fisiológicamente un paciente, pero que no sea vital, aplicará la clase IIa.
“Los programas informáticos destinados a observar procesos fisiológicos se clasifican en la clase IIa”
En el caso de un software que se utilice en la atención de emergencia, en procesos de gestión de la anestesia, o en cuidados intensivos, aplicarán la clases IIb / III.
“un deterioro grave del estado de salud de una persona o una intervención quirúrgica, en cuyo caso se clasifican en la clase IIb”
En el caso de un software que se utilice para apoyar a la concepción, realizando recomendaciones o asistiendo en la detección del embarazo, aplicará la clase I.
“Todos los demás programas informáticos se clasifican en la clase I”
Una vez tenemos identificados los riesgos que aplican al software de salud que estamos desarrollando y su clasificación, el equipo de investigadores y desarrolladores deben determinar por un lado los requisitos de ciberseguridad y de usabilidad que les son de aplicación. Deberá de trazarse un plan para verificar que no hayan puntos que dificulten la usabilidad en ningún aspecto de la interfaz, los cuales puedan llevar a errores de uso que potencialmente afecten a la seguridad de su producto y su funcionamiento.
Por otro lado, el equipo de investigadores y de desarrolladores deberán determinar todos los otros requisitos regulatorios que aplican al software de salud desarrollado. Estos pueden ser los relacionados con la gestión de riesgos, de gestión de calidad, de ciclo de vida del software, así como de evaluación clínica. Habrá que planificar y realizar estudios para demostrar a las autoridades que se han cumplido con los objetivos regulatorios de forma objetiva.
Teóricamente, la regulación no obliga a realzar estos estudios de una forma determinada. A la práctica, siempre que exista una norma técnica (comunmente denominada estándar), esta deberá de seguirse. Las normas técnicas o ISOs se publican de forma recurrente y pueden llegar a quedar armonizadas al marco legal europeo. Esto sucede cuando la comisión europea emite un mandato para que los organismos de estandarización europeos (CEN y CENELEC) revisen los estandares para garantizar que quedan cubiertos.
| Requerimiento | Estándar | Título |
| Quality Management System (QMS) | EN ISO 13485:2016 / AC:2018 | Medical Devices – Quality Management Systems – Requirements for regulatory purposes |
| Risk Management System (RMS) | EN ISO 14971:2019 | Medical Devices – Application of risk management to medical devices |
| IEC / TR 80002.1 | Medical device software – Part 1: Guidance on the application of ISO 14971 to medical devices | |
| Software Lifecycle | EN 62304:2006 / AC:2008 | Medical device software – Software life-cycle processes |
| IEC 82304-1:2016 | Health software – Part 1: General Requirements for product safety | |
| ISO / IEC 14764:2006 | Software Engineering – Software Life Cycle Processes – Maintenance | |
| Clinical Evaluation | MDCG 202-1 | Guidance on clinical evaluation (MDR) / Performance evaluation (IVDR) of medical device |
| IMDRF / SaMD WG / N41 FINAL:2017 | Software as a Medical Device (SaMD): Clinical Evaluation | |
| Cybersecurity | ISO / IEC 27000:2018 (en) (series) | Information Technology – Security techniques – Information security management systems |
| MDCG 2019-16 rev.1 | Guidance on cybersecurity for medical devices | |
| IMDRF / CYBER WG /N60FINAL:2020 | Principles and Practices for Medical Device Cybersecurity | |
| Usability | IEC 62366-1:2015 | Medical devices – Application of usability engineering to medical devices |
| ISO 9241-210:2010 | Ergonomics of human-system interaction – Human-centered design for interactive systems |
La normativa aplicable es la MDCG 2019-16, sobre la cual aplicarán también diferentes legislaciones. Estas giran principalmente entorno a la protección de datos y otras ciber-amenazas. Además, las medidas de mitigación de riesgos de seguridad cibernética pueden crear riesgos de seguridad adicionales. En dicho caso, estos también deben tenerse en cuenta en la gestión de riesgos de seguridad. Encontrarás mucha más información relacionada con la ciberseguridad y las leyes aplicables en este otro artículo.

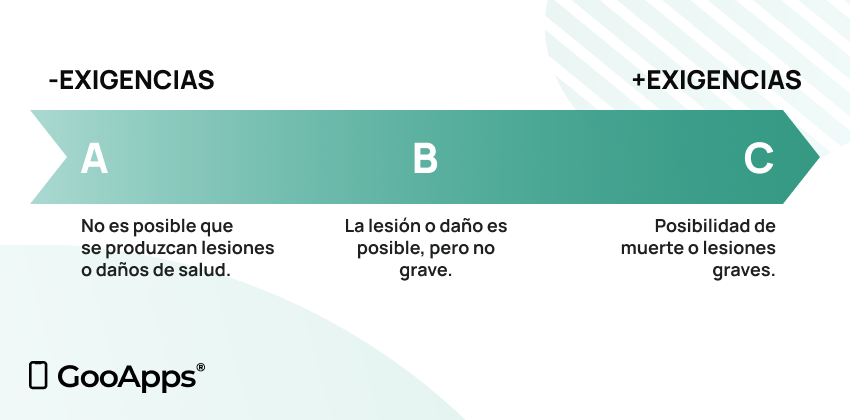
IEC 62304 identifica tres clases de seguridad para el software de dispositivos médicos, en función de la posibilidad de lesiones o daños para sus usuarios:
Será importante alinear el plan de desarrollo entorno a esta norma, la cual ofrece un marco de buenas prácticas para el desarrollo de software médico. Si el software a desarrollar es de clase C, entonces tendremos que cumplirla de forma estricta. Resultará esencial documentar todos los procesos en base a la norma IEC 62304.
Otro requisito regulatorio imprescindible a tener en cuenta para el software médico es la gestión de riesgos. Comprende todo lo relacionado con el análisis, la evaluación y el control de los riesgos que pueden derivar del desarrollo de software de salud. Estos requisitos regulatorios vienen recogidos principalmente en la ISO 14971 y la IEC 62304.
Este paso supondrá la puesta en desarrollo y producción del software médico. En este artículo encontrarás mucha más información al respecto y una guía paso a paso de cómo desarrollar software de salud.

Llegados a este punto, habremos realizado todo el desarrollo de software médico teniendo en cuenta todas las regulaciones que le eran de aplicación. En caso de que existieran normas técnicas, las debemos haber comprado, estudiado y aplicado.
Realizaremos entonces todas las verificaciones y validaciones correspondientes para posteriormente redactar la documentación técnica con los siguientes puntos:
Deberemos de establecer procesos a través de los cuales podamos asegurarnos de que cumplimos con los criterios de gestión de calidad. Esto lo haremos trazando roadmaps y revisiones críticas de los planes de desarrollo.
Una buena estrategia será realizarnos constantemente la pregunta ¿Qué me estoy dejando? De este modo, nos estaremos asegurando de estar cumpiendo la normativa técnica y los requisitos regultarorios, evitando perder así tiempo y dinero.
Último paso del proceso en el que recogeremos toda la documentación técnica y de gestión de calidad para enviársela al organismo notificado. Se realizará entonces el procedimiento de evaluación de la conformidad de nuestro software médico.
Depende. Si te planteas no hacer estudios propios tienes que poder argumentar que hay suficiente evidencia clínica de la misma tecnología de la que está hecha el producto. Es decir, para el mismo grado de complejidad, para su mismo uso previsto, y con las mismas reivindicaciones de uso clínico.
Para productos innovadores será difícil poder apoyarte en evidencias clínicas existentes, ya que tiene poco sentido desarrollar un producto que no esté diferenciado de la competencia. Esto será particularmente crítico para productos de clase III, aunque incluso para un caso I tendrás que hacer estudios propios.
No está tabulado. Se debe diseñar un estudio clínico en el que se plantean unas variables primarias y secundarias para refutar la hipótesis. En función del intervalo de confianza que se suponga mínimo se van a requerir más o menos volumen de pruebas para demostrar científica y clínicamente el uso previsto. Una referencia serán publicaciones de otros softwares que estén en la literatura para determinar si resulta suficiente con 50, 100 o 1000 pacientes.
En este caso no estaríamos hablando de software médico, ya que para que sea considerado como tal debe procesar los datos de algún modo.
La inteligencia artificial implica una serie de procesos de entrenamiento y de evolución del software con un cierto grado de libertad. Habrá que poder demostrar al organismo notificado que el software cumple con la normativa a medida que va evolucionando, ya que se exigirá un control sobre la evolución del software que garantice su perfil beneficio-riesgo.
El equipo de desarrolladores de GooApps es especialista en el desarrollo de software médico y apps de salud. Te ayudamos a determinar la n0rmativa y regulaciones que aplican al producto que se quiere lanzar al mercado, acompañándote en todo el proceso. Trabajamos siguiendo la norma EN 62304:2006 / AC:2008, así como la norma ISO / IEC 14764:2006 en proyectos con mantenimiento. ¡Contacta con nosotros si tienes cualquier duda!
Con el apoyo de:
![]()
Completa el formulario y GooApps® te ayudará a encontrar la mejor solución para tu organización. ¡Contactaremos contigo muy pronto!





